
2023 Bruins-In-Genomics Summer Undergraduate Research Program
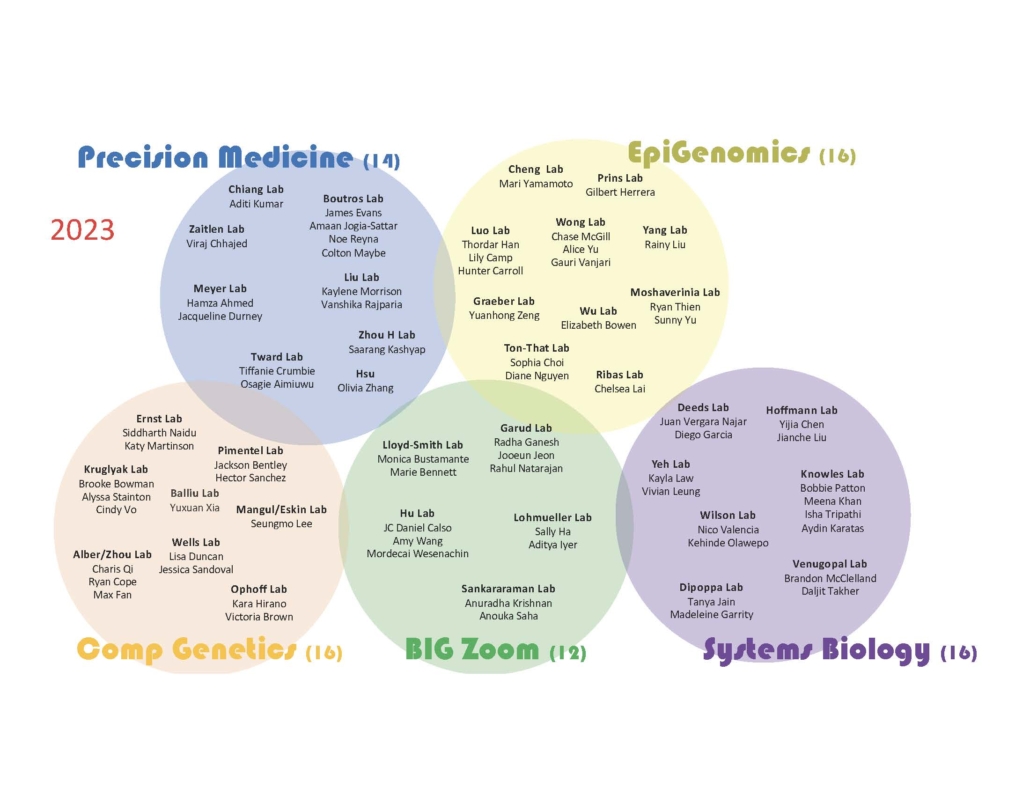
2023 B.I.G. Summer Participants
| Lab PIs | Mentors | Students |
|---|---|---|
| BRUNILDA BALLIU | Yuxuan Xia | |
| PAUL BOUTROS | Zhuyu Qiu & Jee Han | James Evans |
| Zhuyu Qiu & Jee Han | Amaan Jogia-Sattar | |
| Roni Haas & Nicholas Wang | Colton Maybee | |
| Roni Haas & Nicholas Wang | Noe Reyna | |
| GENHONG CHENG | Mari Yamamoto | |
| JEFF CHIANG | Aditi Kumar | |
| ERIC DEEDS | Timothy Hamilton | Diego Garcia |
| Timothy Hamilton | Juan Vergara Najar | |
| MARIO DIPOPPA | Madeleine Garrity | |
| Tanya Jain | ||
| JASON ERNST | Emily Maciejewski | Katy Martinson |
| Emily Maciejewski | Siddharth Naidu | |
| NANDITA GARUD | Ricky Wolff & Michael Wasney | Radha Ganesh |
| Ricky Wolff & Michael Wasney | Jooeun Jeon | |
| Ricky Wolff & Michael Wasney | Rahul Natarajan | |
| THOMAS GRAEBER | Yuanhong Zing | |
| ALEXANDER HOFFMANN | Xiaolu Guo | Yijia Chen |
| Helen Huang | Jianche Liu | |
| WILLIAM HSU | Anil Yadav | Olivia Zhang |
| JIMMY HU | Leah Ye | JC Daniel Also |
| Leah Ye | Yuchen (Amy) Wang | |
| Leah Ye | Mordecai Wesenachin | |
| BEN KNOWLES | Aydin Karatas | |
| Meena Khan | ||
| Bobbie Patton | ||
| Isha Tripathi | ||
| LEONID KRUGLYAK | Joshua Bloom | Brooke Bowman |
| Joshua Bloom | Alyssa Stainton | |
| Chantle Swichkow | Cindy Vo | |
| HONGHU LIU | Ha Luong and Jie Zhang | Kaylene Morrison |
| Jie Shen | Vanshika Rajparia | |
| JAMIE LLOYD-SMITH | Celine Snedden | Marie Bennett |
| Celine Snedden | Monica Bustamante | |
| KIRK LOHMUELLER | Xinjun Zhang | Sally Ha |
| Xinjun Zhang | Aditya Iyer | |
| CHONGYUAN LUO | Matthew Heffel | Lily Camp |
| Matthew Heffel | Hunter Carroll | |
| Cuining Liu | Thordar Han | |
| SERGHEI MANGUL | Vanuri Sarwal | Seungmo Lee |
| AARON MEYER | Jackson Chin | Hamza Ahmed |
| Jackson Chin | Jacqueline Durney | |
| ALIREZA MOSHAVERINIA | Weihao Yuan | Sunny Yu |
| ROEL OPHOFF | Marcelo Francia & Lingyu Zhan | Victoria Brown |
| Marcelo Francia & Lingyu Zhan | Kara Hirano | |
| HAROLD PIMENTEL | Albert Xue | Jackson Bentley |
| Albert Xue | Hector Sanchez | |
| ROBERT PRINS | Lu Sun | Gilbert Herrera |
| ANTONI RIBAS | Nataly Naser Al Deen | Chelsea Lai |
| SRIRAM SANKARARAMAN | Ulzee An | Anuradha Krishnan |
| Ulzee An | Anouka Saha | |
| HUNG TON-THAT | Dana Franklin & Yi-Wei Chen | Sophia Choi |
| Dana Franklin & Yi-Wei Chen | Diane Nguyễn | |
| DANIEL TWARD | Gary Zhou | Osagie Aimiuwu |
| Bryson Gray | Tiffanie Crumbie | |
| SHARMILA VENUGOPAL | Brandon McClelland | |
| Daljit Takher | ||
| MICHAEL WELLS | Alexander Dai | Lisa Duncan |
| Rachel Fox | Jessica Sandoval | |
| JENNIFER WILSON | Kehinde Olawepo | |
| Nico Matthew Sales Valencia | ||
| DAVID WONG | Irene Choi | Chase McGill |
| Irene Choi | Gauri Vanjari | |
| Irene Choi | Alice Yu | |
| LILY WU | Elizabeth Bowen | |
| XIA YANG | Michael Cheng | Ruoshui (Rainy) Liu |
| PAMELA YEH | Wilmer Amaya-Mejia | Kayla Law |
| Wilmer Amaya-Mejia | Vivian Leung | |
| BO YU | Weihao Yuan | Ryan Thien |
| NOAH ZAITLEN | Viraj Chhajed | |
| HONG ZHOU | Saarang Kashyap | |
| JASMINE ZHOU | Ye Wang | Ryan Cope |
| Ye Wang | Max Fan | |
| Ye Wang | Charis Qi |
2023 B.I.G. Summer Poster Abstracts
AHMED, DURNEY: Unraveling Transcriptomic Mechanisms in Persistent Methicillin-Resistant Staphylococcus aureus Bacteremia Through Tensor-Based Data Integration
Unraveling Transcriptomic Mechanisms in Persistent Methicillin-Resistant Staphylococcus aureus Bacteremia Through Tensor-Based Data Integration
HAMZA AHMED¹, JACQUELINE DURNEY¹, Jackson Chin², Aaron Meyer²
1 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2 Department of Bioengineering, Samueli School Of Engineering, UCLA
Methicillin-resistant Staphylococcus aureus (MRSA) bacteremia is a life-threatening infection with a high mortality rate even when treated with appropriate antibacterial therapies. Non-β-lactam antibiotics often fail to fully resolve infections in vivo even when susceptibility is shown in vitro, leading to persistent MRSA bacteremia. The host-pathogen interactions involved in MRSA bacteremia persistence remain poorly understood; to address this, we integrated host transcriptional and proteomic data from patients with MRSA bacteremia using a tensor-based decomposition approach. The large number of assayed genes, however, requires additional processing to recognize transcriptomic patterns. Application of dimensionality reduction including UMAP and Leiden Clustering to the resulting tensor components revealed underlying transcriptomic patterns. Interpretation of the underlying transcriptomic patterns highlight that interferon responses and transcription regulation are key determinants of MRSA persistence outcome. Our results thus shed light on the molecular mechanisms contributing to persistent MRSA bacteremia.
Ahmed:DurneyAIMIUWU: Using simulated data to train convolutional neural networks for image registration
Using simulated data to train convolutional neural networks for image registration
OSAGIE K. AIMIUWU1,2, Daniel Tward3
1 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2 University of North Carolina, Chapel Hill
3 Department of Computational Medicine, Department of Neurology, UCLA
Aligning diverse medical images to the same coordinate system is crucial in subsequent analysis, and has previously been accomplished using convolutional neural networks. The task is complicated by varying contrasts or deformations, and requiring extensive training data. We developed a framework for generating simulated data for training. Mouse brain images were sampled, subjected to random coloring, and underwent random affine transformation. Our network was trained using PyTorch and the Adam optimizer while varying depth, number of features, and learning rate. The best performing network on our validation set had six convolutional and one linear layer, 64 features, a learning rate of 0.001, and a RMSE of 11.246 degrees for rotation and 1.378 pixels for translation on our test set. This model can be used to quickly align images of different modalities or stains and was developed using a novel simulated data framework that generalizes to other applications.
AimiuwuBENNETT, BUSTAMANTE: Correlating Invasive and Non-invasive Methods of Viral Detection in Non-human Primates Infected with SARS-CoV-2
Correlating Invasive and Non-invasive Methods of Viral Detection in Non-human Primates Infected with SARS-CoV-2
MARIE BENNETT1, MONICA BUSTAMANTE1, Celine Snedden2, James O. Lloyd-Smith2,3
1 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2 Department of Ecology and Evolutionary Biology, UCLA
3 Department of Computational Medicine, UCLA
To monitor viral infections and assess within-host viral distribution, animal challenge experiments rely on invasive (e.g., nasal tissue) and non-invasive (e.g., nasal swab) sampling methods. Invasive tissue samples provide valuable information directly from the target location at necropsy, while non-invasive samples are more convenient and can be obtained multiple times throughout infection. Despite the importance of these sample types, the relationship between their outcomes remains unknown. In this study, we used linear regression to assess the correlations between invasive and non-invasive sampling methods across the respiratory and gastrointestinal systems, using a comprehensive database of viral load and infectious virus measurements from non-human primates experimentally infected with SARS-CoV-2. Our analyses offer crucial insights into when non-invasive sampling methods give similar results as invasive samples, including how this may vary over the course of an infection. This could promote animal welfare in future animal challenge experiments by reducing the amount of animals euthanized.
BENTLEY, SANCHEZ: Using dotears to build causal Gene Regulatory Networks on Genome-wide Perturb-seq
Using dotears to build causal Gene Regulatory Networks on Genome-wide Perturb-seq
JACKSON BENTLEY1, HECTOR SANCHEZ1, Albert Xue2,3,4, Harold Pimentel2,3,4
- BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
- Pimentel Lab, Institute for Quantitative and Computational Biosciences, UCLA
- Department of Computational Medicine, David Geffen School of Medicine, UCLA
- Department of Human Genetics, David Geffen School of Medicine, UCLA
Gene regulatory networks describe the causal relationships between multiple different genes and their expression levels, which makes the networks important to understanding the genotype to phenotype landscape more clearly. Furthermore, recent methodological advances like dotears allow inference of causal regulatory networks from interventional data. High-throughput CRISPR screen provides interventional data by measuring gene expression after suppression of individual genes. We apply dotears to a single cell genome-wide CRISPR screen to uncover regulatory relationships in lipid pathways. We applied dotears to a set of 13 genes relevant to cholesterol levels, and found 8 relationships. In particular, multiple relationships were found originating from the genes TCP-1 and CCT3, indicating that dotears correctly identified the two genes as chaperone molecules.
dotears_perturbseq_BOWEN: Interpreting Biological Assays Using Automated Computational Methods
Interpreting Biological Assays Using Automated Computational Methods
ELIZABETH BOWEN1, Lily Wu2, Moe Ishihara3
1 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2 Department of Molecular and Medical Pharmacology, UCLA
3 Molecular and Medical Pharmacology PhD program, UCLA
Wound healing and Transwell Boyden chamber-based migration/invasion assays are crucial for quantifying cell invasiveness and motility, but manual analysis is time-consuming, lacks standardization, and is prone to errors. In this project, we have created an open-source program called AssayAnalyze which automates the quantification process, providing reliable and streamlined analysis. It accurately measures the cell-free region in scratch wound healing assays and recognizes and counts cells in Transwell assays through an intuitive user interface. . We evaluated its performance against manual analysis in a forthcoming study on Von Hippel Lindau negative tumor cell-derived exosomes in metastatic kidney cancer. This comparison confirmed its efficacy, reducing analysis time while maintaining accuracy. AssayAnalyze significantly improves efficiency, accuracy, and reliability, streamlining data acquisition in cancer research and cell biology studies. AssayAnalyze offers a valuable solution for enhanced analysis and insights into tumor cell behavior in cancer progression.
BROWN, HIRANO: Characterizing short tandem repeat expansions in Tourette syndrome
Characterizing short tandem repeat expansions in Tourette syndrome
VICTORIA BROWN1, KARA HIRANO1, Lingyu Zhan2, Marcelo Francia3, Tourette Association of America International Consortium for Genetics (TAAICG), Roel A. Ophoff2
1 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2 Center for Neurobehavioral Genetics, Semel Institute for Neuroscience and Human Behavior; David Geffen School of Medicine, UCLA
3 Department of Neuroscience, David Geffen School of Medicine, UCLA
Expansions of short tandem repeats (STRs) have been implicated in a number of brain disorders. Tourette syndrome (TS) is a highly heritable neurodevelopmental disorder with an unknown role of STRs in disease susceptibility. We examined 38 previously identified pathogenic STRs in whole-genome sequence data of 497 trios (n=1497 subjects) with TS-diagnosed probands using the ExpansionHunter tool. Statistical analysis of these loci revealed no evidence for enrichment of expanded repeats in TS. However, we uncovered a consistent overestimation of repeat lengths on sex chromosomes in females compared to males by ExpansionHunter. Additionally, classifying repeats based on reported pathogenicity ranges revealed multiple pathogenic alleles in our cohort, with the majority of observations at TCF4, a locus previously implicated with schizophrenia and bipolar disorder through genome-wide association studies. Further genome-wide investigation of STR loci is needed to examine their causal relationship to TS and to explore the contribution of novel expansions.
Brown:HiranoCALSO, WANG, WESENACHIN: Dissecting the Signaling Interactions and Cell Lineages in the Developing Mandibular Mesenchyme Using Single-Cell RNA-Seq Analysis
Dissecting the Signaling Interactions and Cell Lineages in the Developing Mandibular Mesenchyme Using Single-Cell RNA-Seq Analysis
JC CALSO1, YUCHEN WANG1,2, MORDECAI WESENACHIN1, Leah Ye3, Jimmy K. Hu3
1 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2 Department of Biology, Williams College
3 Division of Oral and Systemic Health Sciences, UCLA School of Dentistry
The functionality of the mammalian mandible depends on the correct formation of multiple tissue types during embryonic development. However, the genetic and signaling mechanisms that determine cell fates and organize cells into functional structures in the developing mandible remain poorly understood. Here, we analyzed scRNA-seq data from embryonic day 12 mouse mandibular mesenchymal cells using clustering, graph network analysis, and different packages available through R and Python to interrogate cell compositions, signaling interactions and differentiation trajectories. We identified major signaling pathways related to osteogenesis as well as the key cell lineages of the different cell types present in the mesenchyme. Our result lays the groundwork for understanding the development of the mandibular mesenchyme. Combined with our previous characterization of the oral epithelium, this work will provide a rich resource for the comprehensive genetic profiling of the developing mandible and inform future efforts to better understand pediatric craniofacial disorders.
CAMP: Ganglionic Eminence Sequencing Unveils Cortical and Hippocampal Inhibitory Neuron Origins
Ganglionic Eminence Sequencing Unveils Cortical and Hippocampal Inhibitory Neuron Origins
LILY CAMP1,2, Matthew Heffel3,4, Cuining Liu3, Chongyuan Luo3
1 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2 Northwestern University
3 Department of Human Genetics David Geffen School of Medicine, UCLA
4 Bioinformatics Interdepartmental Program, UCLA
The ganglionic eminence (GE) plays a critical role in brain development, yet its molecular mechanisms remain elusive. While excitatory neuron (ExN) development in the hippocampus (HPC) and prefrontal cortex (PFC) has been profiled, incorporating the GE allows us to find inhibitory neuron progenitors, since inhibitory neurons (InN) migrate from the GE to other regions. Thus, we combined mid-gestation HPC, PFC and GE gene expression data to reveal genes involved in the developmental trajectories of InN. Here we perform pseudotime analysis on single-nucleus RNA-seq data from the NeMO archive in Scanpy and reveal region and cell type specific markers in InN differentiation. For radial glia, VIM was expressed everywhere, PTN and FABP5 were expressed for the hippocampus and cortex, and NNAT and NTRK2 for the GE. We found three paths of early-stage InN in the GE where TPX2 and SOX4 were expressed. These findings can provide insight to neurodevelopment disease mechanisms.
Camp_Poster_BIGSummerCARROLL: Gaussian Mixture Variational Autoencoder for Imputation and Batch Correction of DNA Methylation Data
Gaussian Mixture Variational Autoencoder for Imputation and Batch Correction of DNA Methylation Data
HUNTER CARROLL1,2, Matthew Heffel3,4, Cuining Liu3, Chongyuan Luo3
1 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2 University of Colorado, Denver
3 Department of Human Genetics David Geffen School of Medicine, UCLA
4 Bioinformatics Interdepartmental Program, UCLA
Cytosine methylation (mC) is a crucial epigenetic modification regulating gene expression and cellular development. However, mC suffers from high dimensionality when studying multiple CpG sites astride the genome. When analyzing collective age groups of the prefrontal cortex our data suffers from interpolated sources of variation between the high-variance and low-variance features. To model cellular development between sources of variation, like age, we have investigated the use of neural networks as a remedy for information loss. Additionally, using a Gaussian Mixture (GM) model in the latent space of a variational autoencoder may serve to improve clustering, batch effect correction, and imputation by providing non-discrete variational autoencoder latent space parameters. We examine the use of GM Models in the latent space on the adult prefrontal cortex to showcase validity and set the stage for expanding our method to sizable datasets composed of multiple tissues and ages.
Caroll_Hunter_PosterPresentation-reducedCHEN: Multiple Gene Regulatory Strategies Enable Decoding of Transcription Factor Dynamics
YIJIA CHEN1,3, Xiaolu Guo2,3, and Alexander Hoffmann2,3
1 Bruins-in-Genomics Summer Undergraduate Research Program, 2 Institute for Quantitative and Computational Biosciences, and 3 Department of Microbiology, Immunology and Molecular Genetics, University of California, Los Angeles, USA
Prominent stimulus response transcription factors (TFs) exhibit complex dynamic regulation through feedback and feedforward loops. For instance, NFκB and p53 show oscillatory or sustained activities, modulated in amplitude and duration, while ISGF3 displays multiphase activities due to positive feedback. A recent study identified “signaling codons” that capture stimulus-specific information contained in TF dynamics. However, it remains unclear what gene regulatory strategies are required to distinguish the deployment of different signaling codons. Using a two-step chromatin-associated gene expression model, we simulated gene responses to different signaling codons with 2 million parameter sets. Gene response specificity was quantified, revealing high-specificity genes with distinct regulatory strategies. We found that the deployment of activation “Speed”, “Peak”, and “Integral” shared similar regulatory strategies including long mRNA half-life, slow rate to deactivate the poised state, and high TF-responsivity to open or activate the gene promoter. “Duration”, and “Oscillatory Content” were also decoded by similar gene regulatory strategies characterized by short mRNA half-life. Specific parameter sets that describe virtual genes were identified that are capable of providing optimal distinction for the deployment of each signaling codon. Our work sheds light on how genes decode TF dynamics, and provides the basis to interpret experimental stimulus-response RNAseq datasets.
ChenCHHAJED: Leveraging Probabilistic Programming for Flexible Inference in High-Dimensional Statistical Genetics Models
Leveraging Probabilistic Programming for Flexible Inference in High-Dimensional Statistical Genetics Models
VIRAJ CHHAJED1,2, Richard Border 3,4, Noah Zaitlen3,5
1 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2 Henry Samueli School of Engineering, UCLA
3 Department of Neurology, David Geffen School of Medicine, UCLA
4 Department of Computer Science, UCLA
5 Department of Human Genetics, UCLA
Many scientific hypotheses can be formulated in terms of specific parameters in the context of a generative model. However, performing inference about these parameters in large datasets is often a challenging task, frequently requiring bespoke software implementations for every question of interest. This means that much of researchers’ time is spent developing methods for inference rather than addressing questions of direct scientific consequence. In this project, we introduce a novel approach to streamline hypothesis interrogation using NumPyro, a Probabilistic Programming Language (PPL), and efficient Bayesian inference techniques. Our method builds on existing techniques, starting with GWAS and Linear Mixed Effects Model and can be extended to non-infinitesimal and bivariate models, which are particularly relevant for high-dimensional genotype data. By employing PPLs and Hamiltonian Monte Carlo (HMC), we hope to demonstrate that researchers can greatly reduce the time and effort involved in methods development. This approach holds the potential to accelerate scientific discovery, allowing researchers to focus more on hypothesis generation and exploration.
ChhajedCHOI, NGUYEN: Investigation of bacterial microcompartment assembly in Fusobacterium nucleatum
Investigation of bacterial microcompartment assembly in Fusobacterium nucleatum
SOPHIA CHOI1*, DIANE NGUYEN1*, Dana Franklin2, Yi-Wei Chen3, and Hung Ton-That2,3
1Bruins in Genomics, University of California, Los Angeles, California, USA; 2Molecular Biology Institute, University of California, Los Angeles, California, USA; 3Division of Oral & Systemic Health Sciences, School of Dentistry, University of California, Los Angeles, California, USA; *Contributed equally
Fusobacterium nucleatum is an oral commensal but associated with adverse pregnancy outcomes. Preliminary studies revealed that F. nucleatum utilizes ethanolamine from the placenta as nutrients through an ethanolamine utilization (Eut) system promoting preterm birth. This process involves the formation of bacterial microcompartments (BMCs) to compartmentalize ethanolamine metabolism. However, it is unclear whether the BMCs represent a widespread evolutionary adaptive feature. By analyzing Eut orthologs in approximately 69 Fusobacterium genomes from available databases, we found significant variations of Eut determinants among different Fusobacterium species and subspecies, but high conservation in the same taxonomy groups. To study BMC assembly, we constructed a recombinant plasmid expressing potential BMC components (EutL/M1/M2/N), using crossover PCR with specific primers. The generated plasmid were transformed into Escherichia coli DH5α. Once confirmed by DNA sequencing, this plasmid will benefit future studies that examine BMC formation in a heterologous system to determine the essential BMC determinants in F. nucleatum.
Choi:NguyenCOPE, QI: Unraveling the Role of 3D Genome Organization in Modulating DNA Damage Susceptibility and Gene Expression during Chemotherapy
Unraveling the Role of 3D Genome Organization in Modulating DNA Damage Susceptibility and Gene Expression during Chemotherapy
RYAN COPE1, CHARIS QI1, Max Fan1, Ye Wang2,3,4, Frank Alber2, Xianghong Jasmine Zhou3
1BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2Department of Microbiology, Immunology, and Molecular Genetics, UCLA
3Department of Pathology and Laboratory Medicine, David Geffen School of Medicine, UCLA
4Bioinformatics Interdepartmental PhD Program, UCLA
Platinum-based chemotherapy drugs kill cancer cells by damaging DNA. Previous studies have found that the 3D genome structure of cells can influence the distribution of chemotherapy-induced DNA damage. However, the explicit role this regulation plays in drug resistance remains unexplained. Our research explores the relationship between alterations in 3D genome structure of cancer cells and their resistance to platinum-based chemotherapy. By comparing oxaliplatin-resistant and sensitive cells, we observed that changes in DNA damage levels across genomic regions correlate with their nuclear location. Genomic regions near the nuclear speckle displayed reduced damage levels in resistant cells, suggesting that 3D genome adaptations increase resistant cell survival. Additionally, we identified genes with high speckle association frequency that exhibited reduced damage and increased expression levels, implicating them in platinum-drug resistance. This study contributes to our understanding of the interplay between 3D genome structure and chemotherapy resistance, offering potential new avenues for cancer treatment strategies.
COPE:QICRUMBIE: A Deep Learning Technique for Estimating 3D White Matter Fiber Orientation from 2D Histology
A Deep Learning Technique for Estimating 3D White Matter Fiber Orientation from 2D Histology
TIFFANIE CRUMBIE1,4, Bryson Gray2, Daniel Tward2,3
1 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2 Department of Computational Medicine
3 Department of Neurology, UCLA
4 Department of Electrical and Computer Engineering, University of Central Florida
Diffusion Tensor Imaging (DTI) measures 3D macroscopic brain connectivity, but validation against microscopy is challenging due to its 2D nature. Therefore, we propose a Convolutional Neural Network (CNN) framework for estimating 3D fiber orientations in white matter from 2D data. As a proof of concept, we use a 3D human DTI dataset with 8 brains, and train the network to predict “out of plane” information. We built a CNN in pyTorch utilizing U-Net architecture with 7 layers, taking a 2×2 tensor valued image as an input, and predicting a 3×3 image output. We used 6 brains to train our network, and explored parameters using 1 brain for validation. Our model explained 92% of the variance in pixel values on a typical brain slice from our test brain. Our work demonstrates the potential to fill in missing 3D information from 2D data, aiding modern brain atlasing programs and understanding connectivity patterns.
CrumbieDUNCAN: Interrogation of 16p11.2 Region Gene Perturbation
Interrogation of 16p11.2 Region Gene Perturbation
LISA DUNCAN1, Alex Dai2, Michael F. Wells2
1 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2 Department of Human Genetics, David Geffen School of Medicine, UCLA
Copy number variation on chromosomal segment 16p11.2 is the greatest known contributing risk factor for autism spectrum disorder (ASD) and is correlated with other neuropsychiatric disorders, such as bipolar disorder and schizophrenia. Because the effect of 16p11.2 deletion on brain development is not well understood, we explored individual gene function by performing a CRISPR interference screen on 16p11.2 genes in stem cell-derived neural progenitor-like cells (NPCs). We performed Perturb-seq – which captures single guide RNA sequences and single cell gene expression – and analyzed sequencing data using the 10x platform and Cell Ranger pipeline. We identified that experimental improvements will be necessary to optimize the determination of 16p11.2 gene function in NPCs. The results of our analyses will inform future experiments where we aim to identify dysregulated pathways and driver genes of the 16p11.2 region that contribute to ASD and other neuropsychiatric disorders.
LDuncan_poster.BIG2EVANS, JOGIA-SATTAR: Unveiling Survival Signatures: Assessing Contributions of Transcriptional Outliers on Breast Cancer Survival
Unveiling Survival Signatures: Assessing Contributions of Transcriptional Outliers on Breast Cancer Survival
JAMES R. EVANS1,*, AMAAN M. JOGIA-SATTAR1,*, Jee Y. Han2,3,4,5 , Zhuyu Qiu2,3,4,5, Paul C. Boutros2,3,4,5,6
1 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2 Department of Human Genetics, University of California, Los Angeles, USA
3 Department of Urology, University of California, Los Angeles, USA
4 Jonsson Comprehensive Cancer Center, University of California, Los Angeles, USA
5 Institute for Precision Health, University of California, Los Angeles, USA
6 Department of Medical Biophysics, University of Toronto, Toronto, Canada
* equal contributions
Despite their potential as prognostic biomarkers and therapeutic targets, outlier genes with unique expression patterns in cancer have not been comprehensively characterized in the context of patient survival. The integration of outlier gene signatures with clinical measurements enables more nuanced profiling of tumor-specific heterogeneity. Leveraging TCGA RNA-Seq and METABRIC microarray data, we constructed univariate and multivariate Cox proportional hazards models to investigate the impact of outlier genes on patient survival. Our findings shed light on the role of outlier genes as proxies for patient survival, demonstrating their relevance in characterizing breast cancer survival patterns. Exploration of clinical variables in association with outlier genes and their molecular mechanisms can unveil avenues for therapeutic interventions targeting tumor-specific transcriptional variations. Future endeavors include an extension of this methodology across cancer types and advancing pattern identification in pan-cancer outlier contributions for clinical and molecular data.
Evans:Jogia-SattarGANESH, JEON: Why are some fecal microbiome transplants successful and others not?
Why are some fecal microbiome transplants successful and others not?
RADHA GANESH1, JOOEUN JEON1, Michael Wasney2,3, Nandita Garud2,3
1BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2Department of Human Genetics, David Geffen School of Medicine, UCLA
3Department of Ecology and Evolutionary Biology, UCLA
Fecal microbiome transplants (FMTs) are procedures in which gut microbiota from healthy donors are given to diseased patients to treat gastrointestinal disorders. We lack an understanding of what leads to successful colonization by donor gut microbiota, which contributes to FMT success. We hypothesized that successful strain colonization from donor to recipient is positively correlated with the initial community diversity of the recipient gut microbiome, since higher species diversity can potentially open up more ecological niches as per the Diversity Begets Diversity (DBD) hypothesis. We tested our hypothesis on two datasets comprising 34 FMT recipients. We used nucleotide diversity and estimates of strain sharing to assess the relationship between community complexity and colonization success. We found a significant increase in both community and nucleotide diversity in the patient’s gut microbiome shortly after the FMT, while antibiotics significantly decreased community and nucleotide diversity in all patients. We were unable to find a correlation between strain transmission and community diversity prior to FMT. Further research needs to be done in order to reach a conclusion on our hypothesis and understand what factors lead to successful strain colonization from donor to recipient after FMT.
GARCIA, VERGARA NAJAR: Impact of Preprocessing and Cell Exclusion on Stability of scRNA-seq Clustering: Implications for Single-Cell Genomic Analysis
Impact of Preprocessing and Cell Exclusion on Stability of scRNA-seq Clustering: Implications for Single-Cell Genomic Analysis
DIEGO GARCIA1, JUAN VERGARA NAJAR1, Tim Hamilton2, Eric J. Deeds3,4
1 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2 Bioinformatics Interdepartmental Ph.D. Program, UCLA
3 Department of Integrative Biology and Physiology, UCLA
4 Institute for Quantitative and Computational Biosciences, UCLA
Our study explores the effects of preprocessing and clustering on single-cell RNA sequencing (scRNA-seq) data, a revolutionary technology in cellular diversity and disease research. Specifically, this project analyzes whether excluding certain cells, be it the smallest cluster or a random selection, would affect the stability of the clustering results as measured by the Adjusted Rand Index (ARI). We found that the ARI values between clusterings created before and after the removal of certain cells indicated a high divergence between the two clusterings. This finding was consistent across multiple parameter values and datasets analyzed. These discrepancies could lead to errors in cell type identification, amplifying the need for improved clustering and dimensionality reduction algorithms. As we probe the expanding realm of single-cell genomics, our research underscores the need for effective, reliable and interpretable analysis pipelines for single cell data.
Garcia:VergaraGARRITY, JAIN: Increasing specific cell type synaptic strengths drives decorrelation of neural activity in a cortical network model
MADELEINE GARRITY1, 3*, TANYA JAIN1, 2*, Michelle Wu4, Carlos Portera-Cailliau4, Mario Dipoppa4
1 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2 UC Berkeley
3 Rice University
4 Department of Neurobiology, David Geffen School of Medicine, UCLA
*contributed equally
During cortical development, neural activity transitions from highly synchronized to decorrelated firing. The mature decorrelated state is more efficient for neural encoding, but the mechanisms which drive this transition are unclear. Based on biological evidence in rodents, we hypothesized that strengthening connections between specific cell types plays a crucial role in decorrelation. We tested this hypothesis by developing and analyzing biological neural network models. Consistent with our hypothesis, we found that, within a realistic range of network parameters, strengthening synapses from inhibitory neurons cell types caused neural decorrelation. Conversely, other proposed mechanisms, such as decreasing excitability in excitatory cell types, were not sufficient to generate decorrelation. Several results were robust with the addition of spatial connectivity and up-down states to the network. In conclusion, preliminary findings indicate that changing neural inhibition but not excitability alone may drive decorrelation, laying a foundation for research on disruptions to the developmental progression in neural disorders.
JainGarrityPosterHA, IYER: Differences in variant calling methodologies yield different estimations in germline mutation rates
Differences in variant calling methodologies yield different estimations in germline mutation rates
SALLY HA1, ADITYA IYER1, Xinjun Zhang2, Kirk Lohmueller3
1 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2 Department of Human Genetics, University of Michigan
3 Department of Ecology and Evolutionary Biology, UCLA
Germline mutations are the source of several evolutionary events and genetic diseases. Therefore, accurately estimating de novo mutation rates is an important aspect of population genetics. However, due to differences in variant calling methodologies between studies, it can be difficult to provide fully precise estimates of pedigree-based mutation rates. In this work, we aim to benchmark the accuracy and consistency of different variant calling methods and investigate their influence on the discovery of de novo mutations. We applied two commonly used variant calling programs ‒ GATK and FreeBayes ‒ to the same cohort of Mus musculus trios. We further identified de novo mutations from the two sets of genomic variants to emphasize that the choice of bioinformatic tools makes a difference in mutation rate estimation. Our findings substantiate the need for consistent methodology when detecting de novo mutations, laying the groundwork for the development of a standardized pipeline.
HAN: Comparison of Methods for Differential Gene Analysis in Autism Using snRNA Data
Comparison of Methods for Differential Gene Analysis in Autism Using snRNA Data
THORDAR HAN1, Cuining Liu2, Katherine Eyring3, Kevin Abuhanna2, Yi Zhang2, Brie Wamsley3, Daniel H. Geschwind2, 3, Chongyuan Luo2
1 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2 Department of Human Genetics, David Geffen School of Medicine, UCLA
3 Department of Neurology, David Geffen School of Medicine, UCLA
One debate within the single-nucleus RNA sequencing (snRNA-seq) field involves whether to sequence many cells superficially or fewer cells deeply. To investigate this issue, we analyzed two single-nucleus human cortex datasets to detect cell-type-specific differentially expressed genes (DEGs) between autism spectrum disorder (ASD) and control groups. Dataset A profiled 83,958 nuclei (median 2,157 counts/nucleus) using 10X Genomics (Velmeshev et al., 2019). Dataset B profiled 18,666 nuclei (median 96,660 counts/nucleus) using snmCT-seq (Luo/Geschwind Lab). We investigated best practices for integrating these datasets and detecting DEGs. We observed that pseudobulk and cell-level DEG-identification methods have correlated effect estimates, but result in different sets of significant DEGs. Both datasets implicated multiple cell types in ASD with a surprising non-neuronal signature; however, Dataset B yielded a greater number of results, including SFARI-validated autism-related genes that were absent in Dataset A, such as SHANK3, CAMK2A, and UBE3A.
Han_Thordar_Poster-FINALHERRERA: Neoadjuvant anti-PD-1 immunotherapy expands and reinvigorates neoantigen-reactive T cells in recurrent glioblastoma
Neoadjuvant anti-PD-1 immunotherapy expands and reinvigorates neoantigen-reactive T cells in recurrent glioblastoma
GILBERT HERRERA1,2, Lu Sun2, Alexander Lee2, Julio Sanchez2, Roseanna Murray2, Robert Prins2-5
1 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2 Department of Neurosurgery, David Geffen School of Medicine, UCLA
3 Department of Molecular and Medical Pharmacology, David Geffen School of Medicine, UCLA
4 Jonsson Comprehensive Cancer Center, UCLA
5 Parker Institute for Cancer Immunotherapy
While immunotherapies have significantly improved clinical outcomes for cancer patients, their effectiveness against glioblastoma (GBM) brain tumors remains limited. Administration of the immune checkpoint inhibitor anti-PD-1 (aPD1) enhances survival and promotes intratumoral T cell activation, yet disease recurrence remains prevalent. To explore biomarkers of aPD1 response, single-cell RNA and T cell receptor (TCR) sequencing were performed on the tumor-infiltrating immune compartments from 42 recurrent GBM patients. We discovered that aPD1 increases the proportion of a CXCL13+ T lymphocyte population with a distinct neoantigen-specific expression signature. Strikingly, the phenotype sharing the most TCR clones with this cluster shifts from exhausted to stem-like memory T cells after therapy. This suggests that aPD1 enables prolonged functionality of antitumor effector T cells, thereby rationalizing its clinical benefits. From our work, we have identified several candidate TCR sequences that we will clone and insert into T cells to validate their reactivity against patient-derived GBM samples.
HerreraKARATAS: Viral Communities in the Human Gut from Ancient to Modern Epochs
Viral Communities in the Human Gut from Ancient to Modern Epochs
AYDIN KARATAS1, Ben Knowles2,3
1 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2 Institute for Quantitative and Computational Biosciences, UCLA
3 Department of Ecology and Evolutionary Biology, UCLA
The human gut microbiome is central to health, digestion, and immune response. Gut viruses also contribute to human health, but their response to changing human diets from pre-historic to modern timescales has not been thoroughly examined. To address this, we extracted and analyzed viral sequences from publicly available gut microbiome data, including samples from modern-industrial populations, modern-pre-industrial populations, and fossilized feces, known as coprolites. Clustering of viral metabolic gene composition revealed industrial samples exhibited higher homogeneity than pre-industrial and paleofecal samples. This is driven largely by declining frequencies of oxidoreductase genes involved in metabolism of toxins from pre-historic to modern times. Finally, this was associated with an increased temperateness of gut viruses. Altogether, this shows that the modern human gut virome has lost metabolic diversity, is likely less resistant to toxic secondary metabolites, and is more temperate than pre-historic and pre-industrial populations, to deleterious effect on modern health.
KASHYAP: Cryo-Electron Microscopy Characterization of Flagellar Microtubule Doublets in Pathogenic Trichomonas Vaginalis
Cryo-Electron Microscopy Characterization of Flagellar Microtubule Doublets in Pathogenic Trichomonas Vaginalis
SAARANG KASHYAP1,2,5, Alexander Stevens1,2,3,4, Ethan Crofut1,2, Edward Wang1, Patricia Johnson1, Hong Zhou1,2,6
1 Department of Microbiology, Immunology and Molecular Genetics, David Geffen School of Medicine, UCLA
2 California NanoSystems Institute, University of California, Los Angeles, Los Angeles, California, USA
3 Department of Chemistry and Biochemistry, University of California, Los Angeles, Los Angeles, California, USA
4 Biochemistry, Molecular and Structural Biology (BMSB) Graduate Program, UCLA
5 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
6 Electron Imaging Center for Nanomachines, UCLA
Trichomonas vaginalis (Tv) is a parasitic protist and causative agent of Trichomoniasis, the most common non-viral sexually transmitted infection (STI), implicated in perinatal complications and increased rates of HIV transmission. Tv uses its four anterior flagella, composed of 9 outer microtubule doublets (OMD) forming a ring around a central pair of microtubules, to facilitate parasite motility and host cell adherence.The absence of a high-resolution OMD structure hampers our understanding of Tv flagella and its role in drug development. To tackle this challenge, we utilized cryo-electron microscopy (cryo-EM) to image the OMDs of Tv flagella. Here, we performed single particle analysis to resolve a structure of the OMD to 3.8 Å resolution, and identified several uncharacterized internal and external proteins. This work elucidates how specific protein densities provide stability and architecture in the OMD and offers a broader structural basis for flagellar movement in trichomonads.
KashyapKHAN: The Role of Viral Central Carbon Metabolism in Coral Reef Degradation
The Role of Viral Central Carbon Metabolism in Coral Reef Degradation
MEENA KHAN1, Isha Tripathi1, Ben Knowles1,2
1 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2 Department of Ecology and Evolutionary Biology, UCLA
Coral reefs are declining globally. Degraded reefs are often overgrown by bacteria. However, the role of viruses that infect these bacteria, and particularly their metabolic effects, are poorly understood. To address this, we analyzed the frequency of central carbon metabolism genes in viral communities from healthy through degraded reefs, finding that viral metabolism shifts from Pentose Phosphate Pathway and reductive Tricarboxylic Acid Cycle on lytic dominated reefs to Entner-Doudoroff glycolysis on temperate, degraded reefs. We then constructed a compartment model to assess the ecosystem impacts of this metabolic switch. Our model suggests that lytic viral metabolism may lead to further viral production on healthy reefs while temperate viral metabolism might lead to further bacterial overgrowth of degraded systems. Altogether, this implicates viral metabolism in feedback loops that likely enhance ecosystem resilience on healthy reefs and exacerbate the decline of degraded coral reefs.
Meena-Khan-BIG-Summer-PosterKRISHNAN, SAHA: Non-linear linkage patterns improve reference-free imputation for sequencing data
Non-linear linkage patterns improve reference-free imputation for sequencing data
ANURADHA KRISHNAN1, ANOUKA SAHA1, Ulzee An2, Boyang Fu2, Sriram Sankararaman2,3,4
1 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2 Department of Computer Science, UCLA
3 Department of Human Genetics, David Geffen School of Medicine, UCLA
4 Department of Computational Medicine, UCLA
Deep learning-based imputation methods have gained increasing traction in genome-wide association studies to infer missing values in genetic data. Imputation can reveal the mapping of various genes, whole genome resequencing, and how genes influence diseases and traits. However, despite its prevalence in genomics, genotype imputation remains a largely novel technique, and various imputation methods can yield varying results. Linear methods like Beagle5 and Minimac4 have high accuracy rates, but are usually reference based. Other non-linear methods may be reference-free, but contention remains as to which method is the most effective in imputing values. This project examines how reference-free, non-linear machine-learning models, like Logistic Regression and XGBoost, create accurate imputations in chromosome 22. We found that these models improved imputation even in hard-to-predict single nucleotide polymorphisms (SNPs) with lower minor allele frequencies. This proves the effectiveness of non-linear imputation methods and provides a starting point for improvement in generating such models.
KUMAR: Using clustering techniques and polygenic risk scores to classify Type 2 diabetes patients
Using clustering techniques and polygenic risk scores to classify Type 2 diabetes patients
ADITI KUMAR1, Aditya Pimplaskar2, Jeffrey N. Chiang3,4
1 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2 Bioinformatics Interdepartmental PhD Program, UCLA
3 Department of Neurosurgery, David Geffen School of Medicine, UCLA
4 Department of Computational Medicine, David Geffen School of Medicine, UCLA
Type 2 diabetes (T2D) accounts for over 90% of diabetes cases and is heterogeneous in terms of patient’s symptoms, disease progression and response to treatment for the disease. Distinguishing T2D subtypes can be challenging. In contrast to clinical measurements, germline genetic data does not change and can be used to understand T2D heterogeneity. We used k-means clustering to classify UCLA patients into five clusters based on their onset age, BMI and triglyceride, HDL and HBA1C levels. We extracted loci from an existing genome-wide association study (GWAS) and calculated a polygenic risk score (PRS) for each patient. On average patients of higher risk are more likely found in Cluster 5 but the highest risk patients were found in Cluster 4. Understanding the relationship between genetic risk and clinical measurements in T2D patients could improve our ability to understand and manage the disease.
KumarLAI: An analysis of deconvolution methods for spatial transcriptomics data in biopsies of patients with melanoma who received immune checkpoint blockade therapy
An analysis of deconvolution methods for spatial transcriptomics data in biopsies of patients with melanoma who received immune checkpoint blockade therapy
CHELSEA LAI1,2, Nataly Naser Al Deen, PhD2, Egmidio Medina2, Antoni Ribas, MD, PhD2
1 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2 David Geffen School of Medicine, University of California, Los Angeles, Los Angeles, CA1
Spatial Transcriptomics is a cutting-edge molecular profiling method that can profile the transcriptome while preserving the morphological context. However, a limitation of the technology is that each spot (55 µm) may be derived from multiple cells. Fortunately, methods exist to resolve cellular heterogeneity by quantifying the relative contribution from cell types in every spot, termed deconvolution. Melanoma is characterized by a complex tumor microenvironment, thus, studying the cellular mechanisms spatially helps delineate progression and responses to immune checkpoint blockade (ICB) treatment of the malignancy. This study explores and compares several existing deconvolution methods on spatial transcriptome datasets from patients with melanoma who received ICB therapy. We analyzed the deconvolved results alongside the histopathologic annotation, which was used as a reference, to measure the performance of the deconvolution method. Our results reveal that RCTD and CARD performed superior to other methods in resolving cell type deconvolution at the spot level.
LaiLAW: The Co-evolutionary Relationship between the Dark-Eyed Junco and their Avian Haemosporidian Parasites
The Co-evolutionary Relationship between the Dark-Eyed Junco and their Avian Haemosporidian Parasites
KAYLA LAW1, Wilmer Amaya-Mejia2, Pamela Yeh2
1 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2 Department of Ecology and Evolutionary Biology, UCLA
Novel urban environments can alter established host-parasite relationships, resulting in subsequent coevolutionary changes to associated parasites. To determine how environmental changes affect parasites, this study compared infection prevalence of haemosporidian parasites infecting montane (migratory) and urban (resident) dark-eyed juncos (Junco hyemalis) and the relative evolutionary history of this host-parasite relationship. A cladogram was constructed of haemosporidians detected in juncos across southern California using DNA sequences. A tanglegram was constructed to determine the coevolutionary relationship between these parasites and other susceptible avian host species. Infection prevalence was higher at montane sites than urban sites, and our cladogram shows support for resident-specific lineages of Plasmodium sp. (p.p. 96%) distinct from migratory populations. Our tanglegram found lineages of Haemoproteus sp. that have only been reported in the family Passerellidae, suggesting a coevolutionary history. These results indicate that urbanization affects both the prevalence of parasites and their diversification in relation to their hosts.
Law_Kayla_BIGPosterLEE: An integrated framework for structural variant discovery
VISTA: An integrated framework for structural variant discovery
SEUNGMO LEE1*, Varuni Sarwal1*, Jianzhi Yang2, Eleazar Eskin1, Sriram Sankararaman1, Mark Chaisson2, Serghei Mangul2,3
1 Department of Computer Science, University of California Los Angeles, 580 Portola Plaza, Los Angeles, CA 90095, USA
2 Department of Quantitative and Computational Biology, Dana and David Dornsife College of Letters, Arts, and Sciences University of Southern California, Los Angeles, California, 90089, United States
3 Department of Clinical Pharmacy, Alfred E. Mann School of Pharmacy, University of Southern California, 1540 Alcazar Street, Los Angeles, CA 90033, USA
* Denotes equal contribution
Structural variation (SV), refers to insertions, deletions, inversions, and duplications in human genomes. With advances in whole genome sequencing (WGS) technologies, a plethora of SV detection methods have been developed. However, dissecting SVs from WGS data remains a challenge, with the majority of SV detection methods prone to a high false-positive rate, and no existing method able to precisely detect a full range of SVs present in a sample. Here, we report an integrated structural variant calling framework, VISTA (Variant Identification and Structural Variant Analysis) that leverages the results of individual callers using a novel and robust filtering and merging algorithm. In contrast to existing consensus-based tools which ignore the length and coverage, VISTA overcomes this limitation by executing various combinations of top-performing callers based on variant length and genomic coverage to generate SV events with high accuracy. We evaluated the performance of VISTA by using comprehensive gold-standard datasets across varying organisms and coverage. We benchmarked VISTA using the Genome-in-a-Bottle (GIAB) gold standard SV set, haplotype-resolved de novo assemblies from The Human Pangenome Reference Consortium (HPRC), along with an in-house PCR-validated mouse gold standard set. VISTA maintained the highest F1 score among top consensus-based tools measured using a comprehensive gold standard across both mouse and human genomes. VISTA also has an optimized mode, where the calls can be optimized for precision or recall. VISTA-optimized is able to attain the highest precision and sensitivity among other variant callers. In conclusion, VISTA represents a significant advancement in structural variant calling, offering a robust and accurate framework that outperforms existing consensus-based tools and sets a new standard for SV detection in genomic research.
LEE_BIGSUMMER-croppedLEUNG: Characterizing the Gut Microbiome Diversity of Urban and Non-Urban Dark-eyed Juncos (Junco Hyemalis)
Characterizing the Gut Microbiome Diversity of Urban and Non-Urban Dark-eyed Juncos (Junco Hyemalis)
VIVIAN LEUNG1, Wilmer Amaya-Mejia2, Pamela Yeh2
1 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2 Department of Ecology and Evolutionary Biology, UCLA
Urbanization — the process by which areas with greater concentrations of human activities and development are formed — results in environmental changes and subsequent alterations to available resources. Consequently, gut microbial community composition of urban organisms are expected to be affected. Our study examined whether urbanization could affect gut microbiome diversity of dark-eyed juncos. Using iNEXT, alpha diversity metrics were calculated based on MiniOn-sequenced DNA cloacal swab reads of urban and montane junco populations. Urban juncos had higher Shannon diversity index values compared to montane juncos based on the Welch two-sample t-test (t=2.8850, df=11, P=0.0148). Our results suggest that urban-associated resources increase microbial diversity—possibly due to increases in plasticity of junco gut microbiomes correlated with urbanization-based changes in environment, resources and microbial sources. By studying how increases in microbial diversity alter mutualistic relationships between gut microbiomes and their hosts, the regulation of organism adaptation to increasing urbanization could be further understood.
Leung-BIG-Summer-2023-Final-PosterLIU: Assessing the consistency of computational tools for quantifying key antibody repertoire metrics
Assessing the consistency of computational tools for quantifying key antibody repertoire metrics
JIANCHE LIU1, Helen Huang2,3,4, Haripriya Vaidehi Narayanan2,3, Alexander Hoffmann2,3
1 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2 Department of Microbiology, Immunology, and Molecular Genetics (MIMG), UCLA
3 Signaling Systems Laboratory, Institute for Quantitative and Computational Biosciences, UCLA
4 Bioinformatics Interdepartmental PhD Program, UCLA
B-cell undergoes somatic recombination and hypermutation to construct diverse B-cell receptors (BCRs) to bind different antigens. The diversity in these BCR sequences poses challenges in drawing biologically meaningful conclusions, calling for effective computational software. In the past decade, several tools have been developed to assign germline genes, determine complementarity-determining region 3 (CDR3), and characterize sequence mutation frequency and selection landscape. However, no work has been done to benchmark the performance of these tools and guide the selection of software. Here, we implemented a few of the highly cited software (Change-O, MixCR, and Partis) to compare the BCR repertoire between NFκB mutant and wild-type mice. We found that these software packages showed consistency in summarizing clonal diversity and CDR3 length distribution but diverged in quantifying mutation frequency and selection pressure. Our results demonstrate the value of comparing software using real data, and provide insights into software selection in BCR repertoire analysis.
LiuLIU: Cell Type-specific Gene Regulatory Network Atlas: A Repository of cell-type networks with Disease and Pathway Annotation
Cell Type-specific Gene Regulatory Network Atlas: A Repository of cell-type networks with Disease and Pathway Annotation
RUOSHUI LIU1, Michael Cheng2,3, Julie Tran3, Xia Yang2,3,4,5,6
1 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2 Bioinformatics Interdepartmental Program, UCLA
3 Department of Integrative Biology & Physiology, UCLA
4 Institute for Quantitative and Computational Bioscience, UCLA
5 Molecular Biology Institute (MBI), UCLA
6 Brain Research Institute (BRI), UCLA
Gene regulatory networks (GRNs) are fundamental in understanding physiological processes and disease mechanisms. While conventional tissue-level GRN inference lacks cell level resolution to capture disease development, current cell-specific GRN algorithms are suboptimal in accuracy and speed. We developed Single Cell INtegrative Gene regulatory network inference (SCING), as it can accurately capture cell-type characteristics in metabolic diseases. We applied SCING to create a network atlas for different cell types in human and mouse and revealed that the networks exhibit a highly connected architecture adhering to a power-law distribution. To assess functional relevance, we leveraged SCING on RNA-seq data from a non-alcoholic fatty liver disease (NAFLD) study and conducted Key Driver Analysis using a hepatocyte network. The key driver hubs recapitulated known pathways significantly altered in NAFLD, such as transmembrane transport and lipid metabolism. The generated networks offer a valuable, publicly accessible resource for future investigations into single-cell networks and regulatory processes.
Liu_BIG_posterMARTINSON: Imputation of human methylation array based on KNN algorithm
Imputation of human methylation array based on KNN algorithm
KATY MARTINSON¹, Aiden Zhang², Emily Maciejewski³, Jason Ernst⁴
1 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2 Computational and Systems Biology Interdepartmental Program, UCLA
3 Computer Science PhD Program, UCLA
4 Department of Biological Chemistry in the David Geffen School of Medicine, UCLA
Whole-genome bisulfite sequencing (WGBS) is a powerful and expensive tool that provides genome-wide single-base resolution of methylated cytosines. Methylation arrays are a cheaper alternative and are commonly used for cohort and EWAS studies, but they suffer from low CpG site coverage. Imputation of missing CpGs is necessary to meet the same coverage of WGBS data. Using the k-nearest neighbors algorithm (KNN), we can accurately extend methylation arrays using IHEC WGBS methylation data as a reference. We calculated nearest neighbors and distances from the reference and used these to predict methylation values for CpG sites not located on the array. We also transferred these calculations to a different platform, the EPIC BeadChip array. Both studies using the KNN algorithm demonstrated higher correlations to the ground-truth than when compared to a baseline. Using algorithms to impute methylation values rather than depending on WGBS data vastly reduces costs and efforts for EWAS studies.
MartinsonMAYBEE, REYNA: Dissecting the germline variants in breast cancer tumors: Polygenic risk scores alter tumor evolution under a two-hit hypothesis for breast cancer
Dissecting the germline variants in breast cancer tumors: Polygenic risk scores alter tumor evolution under a two-hit hypothesis for breast cancer
COLTON S. MAYBEE1*, NOÉ REYNA1*, Roni Haas2,3, Nicholas K. Wang2,3, Paul C. Boutros2,3,4,5
1 Bruins in Genomics Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2 Department of Human Genetics, University of California, Los Angeles, UCLA Department of Urology, University of California, Los Angeles, USA
3 Jonsson Comprehensive Cancer Center, University of California, Los Angeles, USA
4 Institute for Precision Health, University of California, Los Angeles, USA
5 Department of Medical Biophysics, University of Toronto, Toronto, Canada
* equal contributions
Breast cancer is the leading cause of cancer death worldwide. Previous methods attributed the heritable risk of breast cancer diagnosis to common germline variants quantified using Polygenic Risk Scores (PRS). Previous research shows prostate cancer PRSs are associated with fewer somatic alterations in tumors supporting Knudson’s “two-hit” hypothesis where increased germline risk reduces the needed genomic instability for tumorigenesis. PRSs have also been associated with earlier age at diagnosis, and favorable survival outcomes. Our project seeks to understand if similar associations between breast cancer PRS and tumor molecular features exist. Using tumor samples (n = 1,076) from The Cancer Genome Atlas, we calculated breast cancer PRS using Mavaddat et al.’s PRS313. Then, we identified associations with clinical and molecular features using generalized linear models adjusting for ancestry, age at diagnosis, sex, and existing prognostic clinical factors. Preliminary results reveal no association between PRS and CNAs per driver gene nor between PRS and age of diagnosis.
Maybee:ReynaMCCLELLAND: Astrocyte Reactivity and its Causal Association with Neuroinflammation and Disrupted Autophagy in Neurodegenerative Disease
Astrocyte Reactivity and its Causal Association with Neuroinflammation and Disrupted Autophagy in Neurodegenerative Disease
BRANDON MCCLELLAND1, Mursall Mostamand2, Scott Chandler3, Martina Wiedau4, Sharmila Venugopal4
1 Molecular, Cell, and Developmental Biology Program, UCLA
2 Undergraduate Neuroscience Interdepartmental Program, UCLA
3 Department of Integrative Biology & Physiology, UCLA
4 Department of Neurology, David Geffen School of Medicine
Astrocytes, the most abundant glial cells, play numerous homeostatic and supportive roles in the central nervous system. However, in neurodegenerative diseases, they acquire reactive functional states and expedite cell death by intensifying pathological processes such as neuroinflammation (NI) and disrupted autophagy (DA). Targeting one or more of these processes might provide strategies to delay neurodegeneration. In this work, we combine experimental and computational approaches to clarify the causal associations between astrocyte reactivity (AR), NI and DA involved in neurodegeneration. Computational work involves developing a comprehensive network model of empirically validated causal associations between AR-NI-DA triggers and targets in order to predict key motifs of cell death. Experimental work is testing causation of AR by explicit triggers (e.g., TNF-a) and inhibitors (e.g., mTOR) of NI/DA and during disease development in transgenic mouse models for ALS using quantitative immunocytochemistry and live cell Ca2+ imaging. Together this work will uncover causal pathways of neurodegeneration.
MCGILL, VANJARI, YU: Characterizing mitochondrial cell-free DNA in plasma as a novel liquid biopsy biomarker for oral cancer
Characterizing mitochondrial cell-free DNA in plasma as a novel liquid biopsy biomarker for oral cancer
CHASE MCGILL1, GAURI VANJARI1, ALICE YU1, Irene Choi2, Jordan Cheng2, Neeti Swarup2, David Wong2
1 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2 School of Dentistry, UCLA
Plasma cell-free DNA (cfDNA) has been emerging as a promising source of biomarkers for oncological diseases. In addition to analyzing cfDNA from nuclear origin, reports have shown that mitochondria cell-free DNA (mitcfDNA) is also a viable biomarker for cancer detection. Previously, we demonstrated that using broad-range cell-free DNA Sequencing (BRcfDNA-Seq), which couples low-molecular weight extraction with single-stranded library preparation, reveals the presence of ultrashort single-stranded cell-freeDNA (uscfDNA) in plasma in addition to mononucleosomal cell-free DNA (mncfDNA). We observed that BRcfDNA-Seq enhances the retention of mitcfDNA population, which contains characteristics to be a useful biomarker for cancer detection. Using strand-specific nucleases and library preparation modifications, we deduce that mitcfDNA is made up of a combination of ssDNA and dsDNA fragments. Based on end-motif analysis, we predict that the nucleases regulating mitcfDNA are different than those of nuclear origin. Additionally, we found that mitcfDNA has higher inherent coverage and G-Quad signatures.
McGill:Vanjari:YuMORRISON, RAJPARIA: Enhancing Dental Care Access: Introducing the Electronic Dental Referral Management System (EDRMS)
Enhancing Dental Care Access: Introducing the Electronic Dental Referral Management System (EDRMS)
KAYLENE MORRISON1, VANSHIKA RAJPARIA1, Honghu Liu2, Jie Shen3
1 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2 Section of Public and Population Health, UCLA School of Dentistry, UCLA
3 Section of Special Patient Care and Maxillofacial Prosthetics, UCLA School of Dentistry, UCLA
Approximately 14% of Californians do not have any form of dental coverage. To alleviate financial barriers and improve access to dental care, we devised an integrated system, the Electronic Dental Referral Management System (EDRMS), to streamline the process of addressing oral health concerns in children from screening through treatment. Using Microsoft Power Platform, we implemented an Entity-Relationship Diagram (ERD) to create a model-driven app. We designed a data model based on the ERD and designed the app’s user interface and business logic. The app operates as follows: after parental consent is asked for an initial screening, the approved children undergo an initial oral health screening; screening results are used to determine treatment urgency; children are referred to providers and a treatment plan is developed. Overall, the EDRMS can significantly improve dental care by streamlining patient referrals, enhancing treatment coordination, and identifying trends to enhance overall oral health outcomes.
morrison:rajpariaNAIDU: Comparing Variant Pathogenicity Scores to Understand Patterns in Scoring Methods via ScoreHMM
Comparing Variant Pathogenicity Scores to Understand Patterns in Scoring Methods via ScoreHMM
SIDDHARTH NAIDU1,4, Luke Li2,3, Jason Ernst2,3
1BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2Bioinformatics Interdepartmental Program, UCLA
3Department of Biological Chemistry, David Geffen School of Medicine, UCLA
4Department of Biology, Department of Computer Science, Duke University
A multitude of scoring methods have been developed to determine the pathogenicity of genetic variants, using structural, probabilistic, and evolutionary considerations to generate a score that quantifies the impact of variants on complex diseases. As different scoring methods may represent diverse aspects of the variant, we attempted to develop an approach that captures the combinatorial patterns of these scores. Here we present ScoreHMM, a hidden Markov model that takes genomic tracks of multiple scores as input and learns states that summarize their patterns. 51 scoring methods were used from dbNSFP, a database developed for prediction and annotation of nonsynonymous SNPs across the human genome. We used the learned states to characterize the similarities and differences in scoring methods. We show that each state potentially harbors unique biological characteristics through analysis of functional enrichments and overlap with de novo variants in autism.
NaiduBIGPosterNATARAJAN: Automatic identification of interactions between human proteins and viruses, bacteria, and malaria from paper abstracts
Automatic identification of interactions between human proteins and viruses, bacteria, and malaria from paper abstracts
RAHUL NATARAJAN1, David Enard2, Nandita Garud3
1 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2 Department of Ecology and Evolutionary Biology, University of Arizona
3 Department of Ecology and Evolutionary Biology, UCLA
Recently more than two thousand human virus interacting proteins (VIPs) and Plasmodium (malaria)- or Piroplasm- interacting proteins (PPIPs) were discovered by manually scanning the literature and/or performing experimental procedures. Viruses and malaria have been shown to drive positive selection in humans, indicative of the burden that pathogens place on humans. However manual literature reviews are unscalable for discovery of additional VIPs, PPIPs, or novel classes of interacting proteins, e.g. bacteria interacting proteins (BIPs). To address this, here we introduce a natural language processing approach trained on abstracts from the NCBI PubMed database to automatically detect abstracts containing evidence for interacting proteins. Our results indicate, due to high AUROC and AUPRC performance on various datasets, that the model can accurately identify novel interactions between human proteins and pathogens. We apply this model to identify novel VIPs as well as 164 BIPs.
NatarajanOLAWEPO: PathFX Analysis Reveals Drug-Phenotype Associations in Neurodegenerative Disorders
PathFX Analysis Reveals Drug-Phenotype Associations in Neurodegenerative Disorders
KEHINDE OLAWEPO, Jennifer L Wilson¹
Department of Bioengineering, University of California, Los Angeles
Body of Abstract: Patients with neurodegenerative diseases have insufficient treatments to manage the debilitating and prolonged symptoms of their diseases, therefore, there are no curative options. Here we used our PPI model, PathFX, to assess the connections between approved drugs and neurodegenerative disease pathways to better understand potential treatment strategies. We identified 8 neurodegenerative phenotypes in the PathFX database and found network associations with 3 of them among approved drugs in DrugBank. Our analysis revealed 2,142 drug-phenotype relationships involving 1,113 unique drugs. Supporting literature validated pathways identified by PathFX, including a genetic association between Bridging Integrator 1 (BIN1) and Alzheimer’s disease. Expanding our study to 202 additional neurodegenerative phenotypes, we found 715 drug-phenotype relationships involving 549 unique drugs. The top-genes list differed from the initial analysis. Our results indicate robust connectivity between druggable targets and neurodegenerative disease pathways, showing potential for treatment strategies. Furthermore, our analysis demonstrated high gene similarity across neurodegenerative diseases.
Olawepo_PosterPATTON: Bacteria, Grazer, and Virus Persistence in a Predator-Prey Model
Bacteria, Grazer, and Virus Persistence in a Predator-Prey Model
BOBBIE PATTON1, Isha Tripathi1, Aydin Karatas1, Ben Knowles1,2
1 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2 Department of Ecology and Evolutionary Biology, UCLA
Microbes are usually killed by either viral infection or zooplankton grazing. However, few theoretical models incorporate viral as well as zooplankton mortality rates in environmental predation dynamics. To address this, we implemented a virus-host-grazer predator-prey theoretical model and determined how often the model led to the survival of these populations, and which parameters led to this outcome. We had expected to see virus, host, and grazer survival in models parameterized with reasonable initial values. Surprisingly, we observed pervasive population collapses under these conditions. As a result, we then sought to understand which parameters led to persistence. Doing so, we found only 186 out of 100,000 permutations with randomly chosen, reasonable parameter values survived 350 generations. Altogether, this suggests that either the theoretical model is overly constrictive, or populations must display highly specific traits in order to persist.
PattonSANDOVAL: Exploring the Impact of Valproic Acid (VPA) on Neural Progenitor Cells (NPCs) through Cell Villages and Differential Gene Expression Analysis
Exploring the Impact of Valproic Acid (VPA) on Neural Progenitor Cells (NPCs) through Cell Villages and Differential Gene Expression Analysis
JESSICA SANDOVAL1, Rachel Fox2, Timothy Derebenskiy2, Dr. Michael F. Wells2
1 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2 Department of Human Genetics, David Geffen School of Medicine, Los Angeles, CA, USA
Valproic Acid (VPA) is a commonly used mood stabilizer and antiepileptic drug. In utero exposure to VPA is associated with increased risk for neurodevelopmental disorders (NDDs). Neural progenitor cells (NPCs) are crucial in neurodevelopment and may be vulnerable to VPA. We sought to determine VPA’s effect on NPCs using cell villages, a method which pools together cell lines in a shared in vitro environment. Stem-cell-derived NPCs from 12 donors were combined in equal proportions, then separated into two villages, treated with VPA or water control. Single-cell RNA sequencing and the Dropulation algorithm were used to profile gene expression and cell donor identity. We performed differential gene expression analysis using Voom and Dream and identified 356 differentially expressed genes that changed in response to VPA. Continued research into the effects of VPA can transform our understanding of NDDs, leading to improved healthcare, early interventions, and better outcomes for individuals with challenging neurodevelopmental conditions.
SandovalTAKHER: Automated Development of Novel Functional Interactomes Across Brain Scale
Automated Development of Novel Functional Interactomes Across Brain Scale
1DALJIT TAKHER, 1Shreyank Kadadi and 2Sharmila Venugopal
1 Computational and Systems Biology, University of California Los Angeles
2 Department of Neurology, David Geffen School of Medicine, University of California Los Angeles
We leveraged published work on empirically validated causal associations between brain biomolecules to automate the development of novel functional interactomes across brain scale (FIABS). FIABS networks are knowledge graphs with nodes representing brain proteins, neurotransmitters etc., connected by directed edges indicating facilitating (+1), repressing (-1) or lack of pleiotropic actions of diverse activators (e.g., neurotransmitters, cytokines), on diverse targets (e.g., synapse-associated, cell-stress proteins). The edge weights are scaled by a priority score using a custom look-up table. We used Python NetworkX module to generate and analyze FIABS networks. We systematically tested the FIABS simulator to generate simpler networks and encoded the degree centrality using a color guide. To enable multidimensional visualization of the causal relationships between activators and targets, we generated alluvial plots using the Seaborn, Matplotlib and Pandas libraries. FIABS simulator provides a novel network neuroscience tool to elucidate integrative functions of the nervous system in health and disease.
Takher-et-al-1THIEN: Cytotoxicity Analysis of Pulp Capping Material on Dental Pulp Stem Cells
Cytotoxicity Analysis of Pulp Capping Material on Dental Pulp Stem Cells
RYAN THIEN1, 2, Liqun Mao3, Bo Yu4
1 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2 Department of Biomedical Engineering, University of Arizona
3 School of Dentistry, UCLA, Los Angeles, CA, USA
4 Division of Regenerative and Constitutive Sciences, School of Dentistry, UCLA
Vital pulp therapies, including direct/indirect pulp capping, are essential to preserve pulpal health during management of deep caries. Resin-modified calcium silicate cements (RMCSCs) constitute recent advancements in pulp capping materials capable of inducing dentin bridge formation and promoting adhesion with overlying resin restorative material. However, leaching of unpolymerized monomers can lead to cytotoxicity in pulp cells. We aim to analyze and compare the cytotoxicity of TheraCal LC and other clinical RMCSC liners on the dental pulp stem cells (DPSCs). To evaluate these cytotoxicities, we developed a model to mimic clinical situations utilizing pre-cured liner discs by co-culturing them with DPSCs and performing MTT assays to assess effects of variables such as curing time and surface area. Our findings demonstrate that for light-cured mineral trioxide aggregate (MTA) and TheraCal LC pulp capping materials, curing time corresponds to cytotoxicity, and shortened curing time corresponds to increased cytotoxicity.
POSTER-RYAN-THIENTRIPATHI: Estimating Virus-Host Encounter Rates using Mass Action vs Agent Based Modeling
Estimating Virus-Host Encounter Rates using Mass Action vs Agent Based Modeling
ISHA TRIPATHI1 , Nickie Yang1, Ben Knowles1,2
1 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2 Department of Ecology and Evolutionary Biology, UCLA
Viral lysis of microbes is a leading cause of microbial death. Lysis is commonly modeled using the Mass Action term, where encounters are proportional to the products of host and viral densities, despite it not being validated empirically or theoretically. To assess the validity of Mass Action in modeling virus-host encounter rates, we implemented dynamical models using ODEs and agent-based models to determine if it accurately estimates virus and host encounters. Doing so, we found that Mass Action overestimates encounters between virus and host populations by nine orders of magnitude (i.e.,1,000,000,000 fold). Further, we found that implementing models without the Mass Action term leads to stability of both hosts and viral populations while including Mass Action leads to oscillations and collapses within the populations. Altogether, this suggests that the Mass Action term is logically problematic and may not reflect findings in the empirical world.
Tripathi
VALENCIA: Using PathFX to Find Novel Drug Pathways to Treat Schizophrenia, Bipolar Disorder, and Major Depressive Disorder/Unipolar Depression
Using PathFX to Find Novel Drug Pathways to Treat Schizophrenia, Bipolar Disorder, and Major Depressive Disorder/Unipolar Depression
NICO MATTHEW SALES VALENCIA1 2; Jennifer L. Wilson Ph.D3
1 University of Guam
2 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
3 Department of Bioengineering, UCLA
Schizophrenia, Bipolar Disorder, Major Depressive Disorder/Unipolar Disorder are commonly comorbid diseases, though they are often treated individually. Combination treatments increase the chance of adverse reactions, begetting the need for single-drugs or lower variation of drugs to treat comorbid conditions. Many, including ourselves have used protein-protein interaction (PPI) network models to model drug effects. We used our PPI method, PathFX, to identify drugs with associations to multiple psychiatric diseases and identified shared proteins in these predictions. We discovered several shared proteins GNB1, POMC, SSTR5, APP, OXT, GRM5, HCRT, NPY, CHRM3, and SST and literature support for their connection to disease. Previous literature has found POMC, GRM5, SST, PDYN, CNR2, and NPY to be associated with these phenotypes. By assessing shared pathways we predicted several single-drug options for comorbid psychiatric treatments, but we require further assessment to validate these predictions as treatments.
ValenciaVO: Identifying outlier yeast strains from a pool of a thousand strains in maltose growth conditions
Identifying outlier yeast strains from a pool of a thousand strains in maltose growth conditions
CINDY VO1,2, Chantle Swichkow2, Heriberto Marquez2, Leslie Alamo2, Joshua Bloom2, Leonid Kruglyak2
1 BIG Summer Program, Institute of Quantitative and Computational Biosciences, UCLA
2 Department of Human Genetics, David Geffen School of Medicine, UCLA
Genetic variation in yeast impacts fitness in various environments. Saccharomyces cerevisiae strains have diversified genetically due to evolution in different settings. To study the genetic differences that contribute to varying fitness, genome-wide association studies (GWAS) or meiotic recombination techniques are employed to map quantitative trait loci (QTL). The aim is to identify strains or genetic variants with enhanced fitness in maltose growth conditions from a pool of one thousand, three hundred, and eight yeast strains. Genomic DNA is extracted from each representative pool, sequenced, and analyzed, using non-negative least squares regression, to calculate genotype frequency differences before and after growth in maltose conditions. The anticipated result is the identification of genetic variations from strains better adapted to maltose conditions. From this information, a pipeline is created to identify strains with higher fitness and outlier strains in different conditions for genetic mapping.
Vo_Cindy_PosterXIA: A Comparison of Statistical Methods for Mapping of Context-Specific eQTLs
A Comparison of Statistical Methods for Mapping of Context-Specific eQTLs
YUXUAN XIA1, Brunilda Balliu2
1 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2 Department of Computational Medicine, UCLA
Most GWAS signals harbor expression quantitative trait loci (eQTLs). Several methods for mapping eQTLs have been proposed but the comparative performance of these methods under different scenarios of specificity of eQTL effects across context remains unexplored, especially in the presence of repeated sampling and missing gene expression data. Here, we performed an extensive simulation study to evaluate four different methods for identifying context-specific eQTLs. We show that methods which explicitly account for the repeated sampling design, like FastGxC and mixed effect models, successfully control the type I error rate and are more powerful than fixed effect models which ignore this design, with or without missing data. Our findings highlight the importance of accounting for repeated sampling when mapping eQTLs.
YAMAMOTO: Investigating a potential universal influenza vaccine targeting HA epitopes
Investigating a potential universal influenza vaccine targeting HA epitopes
MARI YAMAMOTO1, Lulan Wang2, Genhong Cheng2
- BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
- Department of Microbiology, Immunology, and Molecular Genetics, UCLA
In this study, we present a novel design for a potential universal influenza vaccine that targets highly conserved epitopes of Hemagglutinin (HA) across all Influenza A subtypes. Current vaccines primarily offer protection against only a narrow spectrum of Influenza strains. This limitation underscores the need for a comprehensive, broad-spectrum solution. By employing a systematic approach, we identified key B cell, T cell, and Major Histocompatibility Complex (MHC) epitopes within the HA protein. These epitopes were selected based on their high conservation across diverse influenza strains and their key functional roles in the viral life cycle. By leveraging detailed structural-functional analysis, we further refined these epitopes and elucidated their interactions with the immune system. This research aims to open a new frontier in influenza vaccine design, potentially offering universal protection against the highly mutagenic and pandemic-prone influenza virus.
YU: Supramolecular Hydrogels with Superior Dynamics for Enhanced Neurogenesis Differentiation and Cell Adhesion of Dental Pulp Stem Cells
Supramolecular Hydrogels with Superior Dynamics for Enhanced Neurogenesis Differentiation and Cell Adhesion of Dental Pulp Stem Cells
SUNNY YU1, Weihao Yuan2, Alireza Moshaverinia2
1 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2 Weintraub Center for Reconstructive Biotechnology, Division of Advanced Prosthodontics, School of Dentistry, UCLA
Spinal cord injury is a form of nerve damage that impacts up to 500,000 people worldwide and is a major cause of permanent disability. Current nerve regeneration techniques, however, consist of several limitations. Autografts have a lack of donor sites and result in donor-site while nerve conduits are expensive and lack biocompatibility and bioactivity. Herein, we propose a dynamic hydrogel to enhance neurogenesis and expedite nerve regeneration. Our hydrogel employs a 3D polymeric network that mimics the dynamics of natural ECM to allow for better cell adhesion and neurogenic differentiation. We analyzed the properties of the hydrogel through SEM and rheology, and the cell behavior through PCR and immunofluorescence staining. Our results demonstrated that our hydrogel can support a more dynamic network and mechanical response allowing for greater cell adhesion and neurogenic differentiation. Our hydrogel shows the potential to be a valuable resource to assist in nerve regeneration.
Yu_Poster_BIG4236ZENG: Reanalyzing transcriptomic profile of castration-resistant prostate cancer
Reanalyzing transcriptomic profile of castration-resistant prostate cancer
YUANHONG ZENG1,2, Tian He2, Chia-Chun Chen2, Wendy Tran3, Thomas G. Graeber1,5,6,7
- BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
- Department of Molecular and Medical Pharmacology, University of California Los Angeles (UCLA), Los Angeles, CA, USA
- Department of Microbiology, Immunology, and Molecular Genetics, UCLA, Los Angeles, CA, USA
- Jonsson Comprehensive Cancer Center, UCLA, Los Angeles, USA
- Crump Institute for Molecular Imaging, UCLA, Los Angeles, USA
- California NanoSystems Institute, UCLA, Los Angeles, USA
- Metabolomics Center, UCLA, Los Angeles, USA
Castration-resistant prostate cancer (CRPC) often progresses to more aggressive forms that develop resistance to androgen receptor (AR) targeted therapy, including small cell neuroendocrine prostate cancer. Several recent studies have developed preclinical models to understand cancer progression dynamics. Here, we utilized the Transcriptome and Isoform Interpretation of Large-scale sequencing (TOIL) pipeline to reprocess the RNA-seq data from a xenograft model that recapitulates therapy induced lineage plasticity during CRPC progression. Furthermore, we compared this data to other datasets and experimental models, mainly by looking into differentially expressed genes and altered key pathways, as well as by performing principal component analysis (PCA) projections. The goal is to generate a comprehensive analysis on transcriptome profiling across various datasets and preclinical models that will potentially contribute to future research discoveries.
ZengZHANG: Using integrated radiomic and pathomic-based models to predict progression-free survival in early-stage lung adenocarcinoma
Using integrated radiomic and pathomic-based models to predict progression-free survival in early-stage lung adenocarcinoma
TENGYUE ZHANG1, Anil Yadav*2,3, Ruiwen Ding*2,3, Sean Johnson3, Denise Aberle2,3, Ashley Prosper3, and William Hsu2,3
1 BIG Summer Program, Institute for Quantitative and Computational Biosciences, UCLA
2 Department of Bioengineering, UCLA
3 Medical & Imaging Informatics, Department of Radiological Sciences, UCLA
*Equal contributions
Early-stage non-small cell lung cancer (NSCLC) patients have a high recurrence rate within the first five years of surgery, reflecting a need to predict post-surgical recurrence and offer personalized adjuvant therapies. We utilize radiomic and pathomic features to predict progression-free survival within five years of resection in early-stage lung adenocarcinoma, the most common subtype of NSCLC. Using 106 cases from the National Lung Screening Trial dataset, we extracted radiomic features from lung nodules on pre-surgery computed tomography scans, and pathomic features from hematoxylin and eosin-stained whole slide images of the resected tissue. We leveraged hand-crafted and deep features from each modality and used a linear Cox proportional hazards model that accounts for time-dependent risk factors. Our study showed combining radiomic and pathomic features results in a more accurate progression-free survival prediction model (C-index=0.6336) as compared to using radiomic (C-index=0.6117) or pathomic (C-index=0.5838) features alone.
Zhang_Tengyue_Poster